create hw with classes #11
There are no files selected for viewing
This file contains hidden or bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
| Original file line number | Diff line number | Diff line change |
|---|---|---|
| @@ -0,0 +1,185 @@ | ||
| from Bio.Seq import Seq | ||
| from Bio import SeqIO | ||
| from Bio.SeqUtils import GC | ||
| import numpy as np | ||
| import seaborn as sns | ||
| import matplotlib.pyplot as plt | ||
|
|
||
|
|
||
| class Employee: | ||
|
|
||
| def __init__(self, name, age, work_experience): | ||
| self.name = name | ||
| self.age = age | ||
| self.work_experience = work_experience | ||
|
|
||
| def __str__(self): | ||
| return 'Information about employee: {}'.format(self.name) | ||
|
|
||
| def proposed_position(self): | ||
| ''' | ||
| Shows the position the candidate is applying for | ||
| :return: str, position name | ||
| ''' | ||
| if self.work_experience <= 1: | ||
| position = 'Junior DS' | ||
| elif 1 < self.work_experience <= 3: | ||
| position = 'Middle DS' | ||
| else: | ||
| position = 'Senior DS' | ||
| return position | ||
|
|
||
| def proposed_salary(self, position): | ||
| ''' | ||
| Shows salary the candidate wants to have | ||
| :param position: str, name of the position | ||
| :return: int, salary in dollars | ||
| ''' | ||
| if position == 'Junior DS': | ||
| salary = 1000 | ||
| elif position == 'Middle DS': | ||
| salary = 5000 | ||
| else: | ||
| salary = 10000 | ||
| return salary | ||
|
|
||
| def view_info(self): | ||
| ''' | ||
| Shows basic information about candidate | ||
| :return: str with information | ||
| ''' | ||
| position = self.proposed_position() | ||
| salary = self.proposed_salary(position) | ||
| return f"Name: {self.name}\nAge: {self.age} years\nPosition: {position}\nSalary {salary} $" | ||
|
|
||
|
|
||
| class SetModified(set): | ||
|
|
||
| def __init__(self, *args): | ||
| self.positive_numbers = [arg for arg in args if isinstance(arg, (int, float)) and arg > 0] | ||
| super().__init__(self.positive_numbers) | ||
|
|
||
| def add(self, element): | ||
| if isinstance(element, (int, float)) and element > 0: | ||
| super().add(element) | ||
|
|
||
| def update(self, *elements): | ||
| for element in elements: | ||
| self.add(element) | ||
|
|
||
|
|
||
| class RNA: | ||
|
|
||
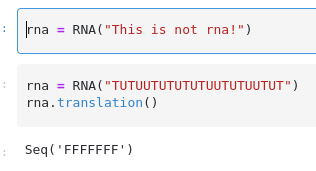
| def __init__(self, rna): | ||
| self.rna = Seq(rna) | ||
|
There was a problem hiding this comment.
|
||
|
|
||
| def translation(self): | ||
| ''' | ||
| Translates the RNA sequence into protein | ||
| :return: protein sequence | ||
| ''' | ||
| return self.rna.translate() | ||
|
|
||
| def back_transcription(self): | ||
| ''' | ||
| Convert RNA sequence into DNA | ||
| :return: DNA sequence | ||
| ''' | ||
| return self.rna.back_transcribe() | ||
|
|
||
|
|
||
| class FastaStats: | ||
|
|
||
| def __init__(self, path): | ||
| self.path = path | ||
|
|
||
| def __str__(self): | ||
| return "Path to the file: {}".format(self.path) | ||
|
|
||
| def count_sequences(self): | ||
| ''' | ||
| Counts number of sequences presented in the file | ||
| :return: int, number of sequences | ||
| ''' | ||
| seq_numb = 0 | ||
| with open(self.path) as f: | ||
| for record in SeqIO.parse(f, "fasta"): | ||
|
Comment on lines
+105
to
+106
There was a problem hiding this comment. Ты считываешь данные в каждой функции. Наверное, лучше выделить это в отдельный метод и считать всё один раз в конструкторе |
||
| seq_numb += 1 | ||
| return seq_numb | ||
|
|
||
| def calc_sequences_length(self): | ||
| ''' | ||
| Calculates length of sequences presented in the file | ||
| :return: list with lengths of sequences | ||
| ''' | ||
| with open(self.path) as f: | ||
| length_ls = [len(record.seq) for record in SeqIO.parse(f, "fasta")] | ||
| return length_ls | ||
|
|
||
| def calc_gc_content_per_seq(self): | ||
| ''' | ||
| Calculates GC content of sequences presented in the file | ||
| :return: list with GC content per each sequence | ||
| ''' | ||
| with open(self.path) as f: | ||
| gc_content_ls = [GC(record.seq) for record in SeqIO.parse(f, "fasta")] | ||
| return gc_content_ls | ||
|
|
||
| def calc_mean_gc_content(self): | ||
| ''' | ||
| Calculates mean GC content over all sequences presented in the file | ||
| :return: int, mean GC content (%) | ||
| ''' | ||
| return np.mean(self.calc_gc_content_per_seq()) | ||
|
|
||
| def calc_k_mers(self, kmer_size): | ||
| ''' | ||
| Counts k-mer over all sequences in the file | ||
| :param kmer_size: int, size of k-mers | ||
| :return: dictionary, where keys - kmers, values - their counts | ||
| ''' | ||
| k_mer_counts = {} | ||
| with open(self.path) as f: | ||
| for record in SeqIO.parse(f, "fasta"): | ||
| for i in range(len(record.seq)-kmer_size+1): | ||
| kmer = str(record.seq[i:i+kmer_size]) | ||
| k_mer_counts[kmer] = k_mer_counts.get(kmer, 0) + 1 | ||
| k_mer_counts = dict(sorted(k_mer_counts.items(), key=lambda item: item[1], reverse=True)) | ||
| return k_mer_counts | ||
|
|
||
| def plot_length_distribution(self): | ||
| ''' | ||
| Plots length distribution for sequences presented in the file | ||
| ''' | ||
| length_ls = self.calc_sequences_length() | ||
| plt.figure(figsize=(10, 8)) | ||
| sns.distplot(length_ls, bins=30) | ||
| plt.title('Length distribution', fontsize=15) | ||
| plt.xlabel('length', fontsize=15) | ||
| plt.ylabel('frequency', fontsize=15) | ||
|
|
||
| def plot_gc_content_distribution(self): | ||
| ''' | ||
| Plots GC content distribution for sequences presented in the file | ||
| ''' | ||
| gc_content_ls = self.calc_gc_content_per_seq() | ||
| mean_gc_content = self.calc_mean_gc_content() | ||
| plt.figure(figsize=(10, 8)) | ||
| sns.distplot(gc_content_ls, bins=30) | ||
| plt.title('GC content distribution', fontsize=15) | ||
| plt.xlabel('GC content, %', fontsize=15) | ||
| plt.ylabel('frequency', fontsize=15) | ||
| plt.axvline(mean_gc_content, color='red', linestyle='--') | ||
|
|
||
| def plot_kmer_distribution(self, kmer_size): | ||
| ''' | ||
| Plots frequency of k-mers over all sequences | ||
| :param kmer_size: int, size of k-mers | ||
| ''' | ||
| k_mer_counts = self.calc_k_mers(kmer_size=kmer_size) | ||
| plt.figure(figsize=(12, 10)) | ||
| plt.bar(k_mer_counts.keys(), k_mer_counts.values(), width=1) | ||
| plt.title('{}-mers distribution'.format(kmer_size), fontsize=15) | ||
| plt.xlabel('{}-mers'.format(kmer_size), fontsize=15) | ||
| plt.ylabel('counts', fontsize=15) | ||
| plt.xticks(fontsize=3, rotation=90) | ||
This file contains hidden or bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
| Original file line number | Diff line number | Diff line change |
|---|---|---|
| @@ -0,0 +1,4 @@ | ||
| biopython==1.79 | ||
| matplotlib==3.1.3 | ||
| seaborn==0.10.0 | ||
| numpy==1.20.0 |
Oops, something went wrong.
Add this suggestion to a batch that can be applied as a single commit.
This suggestion is invalid because no changes were made to the code.
Suggestions cannot be applied while the pull request is closed.
Suggestions cannot be applied while viewing a subset of changes.
Only one suggestion per line can be applied in a batch.
Add this suggestion to a batch that can be applied as a single commit.
Applying suggestions on deleted lines is not supported.
You must change the existing code in this line in order to create a valid suggestion.
Outdated suggestions cannot be applied.
This suggestion has been applied or marked resolved.
Suggestions cannot be applied from pending reviews.
Suggestions cannot be applied on multi-line comments.
Suggestions cannot be applied while the pull request is queued to merge.
Suggestion cannot be applied right now. Please check back later.
There was a problem hiding this comment.
Choose a reason for hiding this comment
The reason will be displayed to describe this comment to others. Learn more.
Записывать это в атрибут не нужно